
Research Projects:

Cystic Fibrosis
Cystic Fibrosis (CF) is a genetic disease caused by molecular variation in the cystic fibrosis conductance transmembrane regulator (CFTR) that manages chloride conductance in the human lung. When deficient in disease the patient develops chronic obstructive pulmonary disease (COPD). We have pioneered the use of GP based computational approaches to understand the impact of variation on the thermodynamics and cell biology of the CFTR protein fold design to discover the root cause of disease in the population. (Bridging Genomics to Phenomics, 2018; CFTR Fold Thermodynamics, 2022; Triangulating Variation, 2022) These results provide mechanistic insights that allow us to discover and define the role of therapeutics in disease management in the clinic as a precision medicine approach .

Alpha-1 Antitrypsin Deficiency
Alpha 1-Antitrypsin Deficiency (AATD) is aging disease of the liver-lung axis (image of liver-lung axis) in response to genetically inherited variants of alpha-1-antitrypsin (AAT), a neutrophil (NE) elastase protease inhibitor. In disease, variants can lead to a gain-of-toxic function aggregation in the liver, preventing the normal secretion of grams per day of AAT to the serum resulting in a loss-of-function disease in the lung and chronic obstructive pulmonary disease (CODP). ( AAT Basal State, 2022; Correction of AATD Hallmarks, 2020) In ongoing efforts, we use GP based SCV relationships to link genotypic diversity in the population to phenotypic diversity in the individual. Our current efforts illustrate the potential for sequence-defined clusters in the AAT protein fold to play very different roles during nascent synthesis in the liver leading to aggregation disease as opposed to loss-of-function in post-liver plasma and lung environments. Using GP we are learning how to manage this diversity through mechanistic description of the proteostasis program that differentially modulates protein fold design in response to a variant- restoring function.
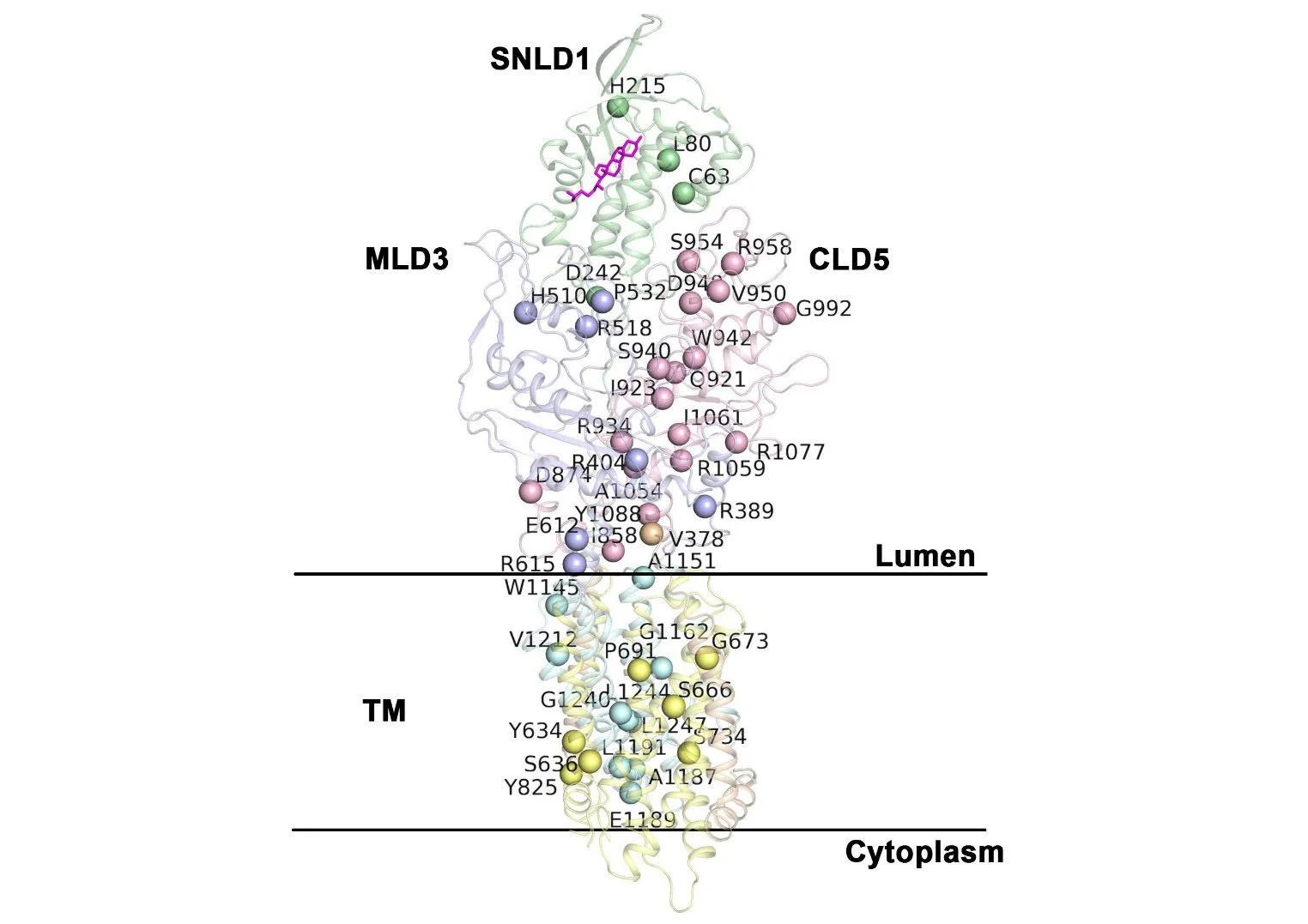
Niemann-Pick CI Disease
Niemann-Pick C1 (NPC1) disease is a rare genetic disorder triggered by mutations in NPC1, a multi-spanning transmembrane protein that is trafficked through the exocytic pathway to late endosomes (LE) and lysosomes (Ly) (LE/Ly) to globally manage cholesterol homeostasis. Defects triggered by >300 NPC1 variants found in the human population inhibit export of NPC1 protein from the endoplasmic reticulum (ER) and/or function in downstream LE/Ly, leading to cholesterol accumulation and onset of neurodegeneration in childhood. Using GP to model genetic diversity in human disease we show quantitatively how the Hsp70 chaperone/co-chaperone system can adjust the spatial covariance (SCV) tolerance and set-points on an amino acid residue-by-residue basis in NPC1 to differentially regulate variant trafficking, stability, and cholesterol homeostasis. (Modulation of the HSP70 Chaperone System, 2020) Moreover, we are actively pursuing the role of epigenetics (Quantitating the epigenetic, 2019) in NPC1 protein fold design to learn how to adjust the SCV tolerance of both proteostatic and epigenetic pathways provide buffering capacity to restore fold function to mitigate systemic and neurological disease in the NPC1 population.

SARS-CoV2
One of the major challenges in understanding variation is its role in host-pathogen relationships. This is particularly exemplified by the recent SARS-CoV-2 worldwide pandemic. We are developing new ways to explore the fast-track evolution in the viral genome on a allele-by-allele basis (~30,000 nucleotide bases) to understand the role of variation in each of the genome encoded genes involved in uptake (Spike), replication (Nsp12 polymerase) (Covariant Fitness Clusters in SARS-CoV-2), packaging (many proteins), and release as a collective to optimize its ability to exploit the human population. The question we are addressing is how the evolving SARS-CoV-2 sequence, encoded through GP based SCV relationships, can generate the successive break-through surges in the context of different lineages including the Alpha, Delta, and Omicron VOCs to continually drive the pandemic. Referred to as the 'Red Queen' effect: “Now, here, you see, it takes all the running you can do, to keep in the same place. If you want to get somewhere else, you must run at least twice as fast as that!”(Through the Looking Glass, Chapter II, Lewis Carroll, 1871). GP allows us to detect underlying spatial and temporal linked (spacetime) probabilistic rules that we posit will provide a broader understanding of the key features in host-pathogen relationships that could enable better therapeutic management. By providing insight into the ‘Red Queen’ effect in which the fast-track variation found in the virus is counter-balanced by reciprocal, yet slower-track, responses by the immune system of the host, as well as therapeutic, social (mask and distancing) and political (lock-down) strategies that factor into virus spread in the host population, we can begin to understand the causal features of disease affecting the individual in age-related manner.

Covariance in Aging Disease
Pulmonary function in response to virus infection, particularly the coronaviruses influenza and SARS-CoV-2, is hypersensitive to aging. This is thought to be a loss in both proteostasis and immune support. As part of an integrated program with Pulmonary Medicine at the Feinberg School of Medicine at Northwestern Medical School, we have found that the small molecule ISRIB, that activates the IRE1/XBP1, a signaling pathway controlling the endoplasmic reticulum (ER) unfolded protein response (UPR), can protect from a loss of pulmonary function during aging. (Lung Injury Induces Alveolar T2 Cell 2021, Resetting Proteostasis with ISRIB, 2021).We are building mass spectrometry descriptions of young and old aging models using influenza challenged mice (Proteostasis and Energetics as Proteome Hallmarks, 2019) as well as clinical data from SARS-CoV-2 variation spanning the world-wide population that can be mined using GP based computational approaches to discover new players and approaches to mitigate the decline in response to virus challenge and extend healthspan of the aging population through proteostasis.

RuBisCo
RuBisCO (ribulose-1,5-bisphosphate carboxylase/oxygenase) is the first step in the process by which atmospheric carbon dioxide (CO2) is converted by plants and other organisms to energy-rich molecules such as glucose in response to energy generated by photosynthesis. As the most abundant protein on earth, we are currently using GP based SCV relationships to assess variation in the genome sequence of RuBisCo found across multiple plant species that is used to optimize RuBisCo fold design for CO2 fixation in different environments. The goal of the project is to provide a GP based universal platform for understanding the genotypic features defining the RuBisCo fold design that will improve plants as a food source and potentially impact climate change through improved global carbon fixation.